Dr. Eduard Matito
Quantum Chemistry
Home
New position for Ukrainian scientists
| Posted on March 28, 2022 at 1:55 PM |
The DIPC offers research visits to Ukrainian scientists (from Ph.D. students to professors) and will cover travel and living expenses (for some months and a limited number of cases) for the candidates selected for the positions described on this webpage.
Elixabete Rezabal and I offer one such position. We are looking for researchers who want to work on a project in the field of ionic liquids. We want to explore IL with large optical responses and analyze various structure-property relationships. We plan two main objectives: (i) analyzing the connection between the electron delocalization in the IL and its first hyperpolarizability and (ii) recognizing chemical building blocks responsible for a sizeable optical response. These results will be used to design tailored IL with enhanced optical properties.
If you are interested in this position or know other candidates that might be eligible for this position, please contact us.
New Ph.D. position in our group!
| Posted on June 18, 2021 at 3:20 AM |
There is a new Ph.D. position in the Quantum Chemistry Development group to work on the design of new density functional approximations. Rubén and I are looking for highly motivated candidates with a taste for method development in electronic structure theory. Candidates from all countries are welcome. Read the ad below and do not hesitate, apply for this position! We are looking forward to work with you!

Development of new density functional approximations
Supervisor(s):
Eduard Matito ([email protected])
Rubén R. Ferradás ([email protected])
Duration: 3 years (possibility of one-year extension based on the availability of funding)
Application Deadline: 31/08/2021
Since its appearance, the density functional theory (DFT)1 has experienced a vast development, establishing nowadays as the most employed tool to tackle many different aspects of the electronic structure of molecular systems. The density functional theory is, in principle, an exact method. It works with functionals, which give the energy or other properties in terms of the electron density n(r). Only a small but essential contribution to the energy, the exchange-correlation (XC) energy, thus far remains unknown as a functional of the density and has to be approximated. In this regard, current density functional approximations are a far cry from predictable, and consequently, they do not show the desirable transferability among properties.
In contrast to the traditional and widespread density functional approximations based on the local expansion of the XC homogeneous electron gas, this Ph.D. project will follow a sort of bottom-up approach in constructing the XC functional. The main goal of this thesis is to develop new density functional approximations that do not rely on the homogenous electron gas, which is the primary physical model behind most current approximations. Two different strategies will be followed: (i) obtaining the XC potential by inverting the Kohn-Sham equation of a simple physical model, (ii) developing correlation functionals from a physical-motivated partition of the pair density.2 The two- electron Harmonium Atom3 (2e-HA) will be the physical model used for the former purpose. The HA resembles an ordinary atom in which the external Coulomb potential is replaced by a parabolic one, which depends on a confinement parameter ω. The project will furnish new DFAs and DFA ingredients that will significantly differ from the existing ones and could be used as a starting point to further developments in the context of orbital-free DFT.
The position is opened for candidates holding a degree in Chemistry, Physics, or Mathematics. They are expected to have a solid background in quantum mechanics, high-level mathematics, and a keen interest in computational chemistry. Good command of English, both verbal and written, is mandatory. Experience with Mathematica® and programming (preferably in FORTRAN or C++) is highly desirable but not required. The successful candidate will join the Quantum Chemistry Development group at Donostia International Physics Center (DIPC), led by Eduard Matito.
The candidate will be provided with ample office space, all the resources needed to carry out the research and will be encouraged to participate in international conferences. Applicants of all genders and ethnicities are welcome. The deadline to start is January 2022. Interested candidates should submit an updated CV and a brief statement of interest to the supervisors’ email listed above. Reference letters are welcome but not indispensable.
1 R.O. Jones, Reviews of Modern Physics 87, 897 (2015)
2 M. Via-Nadal, M. Rodríguez-Mayorga, E. Ramos-Cordoba, E. Matito, J. Phys. Chem. Lett. 10, 4032 (2019)
3 N.R. Kestner and O. Sinanoglu, Physical Review 128, 2687 (1965)
Tuning the Excited State Huckel-Baird Hybrid Aromatic Character
| Posted on February 16, 2021 at 3:45 AM |
The first article of 2021 has been accepted in Angewandte Chemie. The main author is Sílvia Escayola, who is doing her joint PhD between the DIPC (working with Dr. Eduard Matito) and the Universitat de Girona (Prof. Miquel Solà and Dr. Albert Poater). The work is a collaboration between these groups and photophysics group of Dr. Casanova (DIPC) and the group of Henry Ottosson.
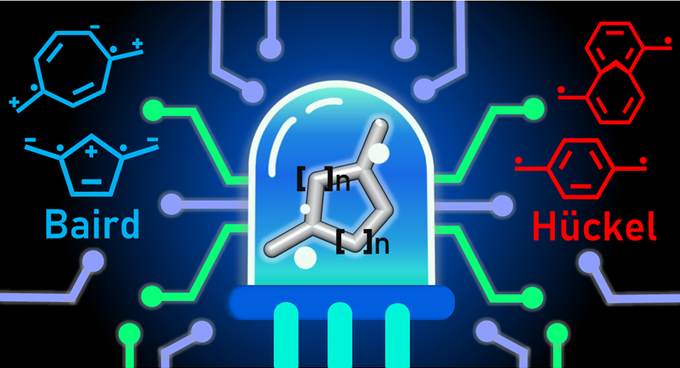
Using time-dependent density functional theory, we considered a series of symmetrically substituted conjugated rings that can generate Baird aromaticity in the lowest-lying excited states. From these results, we establish general guidelines for the rational design of molecules with excited state Hückel or Baird aromaticity in pro-aromatic quinoidal compounds. Namely, two main strategies to promote a high Baird aromatic character of the central ring, are suggested: (i) employing anionic and small conjugated rings with electron donating groups as substituents and small exocyclic groups with electron withdrawing substituents, or (ii) electron deficient conjugated rings with exocyclic electron-donor substitution. Our results also show that some recent experimental interpretation [Kim et al. Nature Commun. 2019, 10, 4983] of the excited state of TMTQ should be revised because low-lying excitations of symmetrically substituted conjugated rings including TMTQ hold very weak charge transfer character.
S. Escayola, C. Tonnelé, E. Matito, A. Poater, H. Ottosson, M. Solà, and D. Casanova. “Guidelines for Tuning the Excited State Hückel-Baird Hybrid Aromatic Character of Pro-Aromatic Quinoidal Compounds” Angew. Chem. Int. Ed. 2021, ASAP (doi: 10.1002/anie.202100261). The paper is also available at chemrxiv.
Three-year postdoc position
| Posted on January 8, 2020 at 10:10 AM |

Density Functional Theory (DFT) is indisputably the most widely employed electronic structure method, used in chemistry, physics, material science, biology and cross-field specialties. DFT is an exact theory but current density functional approximations (DFAs) fail to account for the breaking of chemical bonds, strongly correlated materials, transition metal systems, magnetic properties or dispersion interactions in excited states. These limitations are connected to the three main open challenges in DFT: large self-interaction errors, proper inclusion of nondynamic correlation and description of noncovalent interactions beyond ad hoc semi-empirical corrections. Despite the proliferation of new DFAs, these issues remain unsolved. In this sense, DFT has reached a dead-end point and it needs fresh innovative ideas and ingredients for a new generation of robust all-purpose DFAs to be developed.
This project presents a genuinely new strategy to construct DFAs in which the exchange-correlation functional is decomposed exactly into nondynamic and dynamic correlation components at different interelectronic domains. The separation breaks the complicated exchange-correlation functional into simpler mathematical objects that are easier to treat. The prospective postdoctoral fellow will work on a project that holds the promise of both providing a more accurate and rigorous description of systems within the fields where DFT is currently applied and extending the scope of application to challenging systems.
Candidates should have a strong background on electronic structure theory, and excellent programming skills. Experience on the development of density functional approximations or other computational chemistry methods will be highly appreciated. The position is for one year with the possibility to extend it up to three years.
Interested candidates should submit an updated CV and a brief statement of interest to Dr. Eduard Matito ([email protected]). Reference letters are welcome but not indispensable. Very good communications skills in English are required.
This project has received funding from Spanish Government’s grant program
“Europa Excelencia 2019” under grant number EUR2019-103825 and the DIPC.
DIPC internships to work with us
| Posted on February 6, 2019 at 6:20 PM |
Are you interested in research? Do you want to work on the development of electronic structure methods? The DIPC offers a paid internship in Donostia to work with us.
Quantum mechanics provides the framework to treat molecular systems but its exact application for systems with more than two particles remains elusive. In practice, electronic structure simulations rely on approximate computational methods of variable accuracy. The main obstacle towards the accurate description of molecular systems is the so-called electron correlation. One of the most difficult problems in quantum mechanics is the account of strong correlation. Radical systems, bond activation, magnetic compounds, transition metal complexes, among others, suffer from strong correlation and their correct simulation is hampered by the lack of cost-efficient computational methods.
The cost of the electronic structure methods increases importantly with the system size. Among the computational methods available in the literature, density functional theory (DFT) is the one that provides a best comprise between accuracy and computational cost. Unfortunately, current density functional approximations (DFAs) fail to account for strong correlation and there are thus no cost-efficient methods to study large strongly correlated systems. In this sense, the inclusion of strong correlation in DFAs is one of the greatest present challenges in this field.
In this work, the candidate will explore new models of strong correlation designed in our group. These models will be used to retrieve the strong correlation part of the electron-electron interaction in the context of DFT. The models will be tested on the dissociation of diatomic molecules, radicals systems and transition metal complexes.
The candidate should have a basic knowledge on quantum mechanics (assumed in physics and chemistry BSc. students), and be eager to learn the basics of electronic structure theory and DFT.
See this link for further information on how to apply.
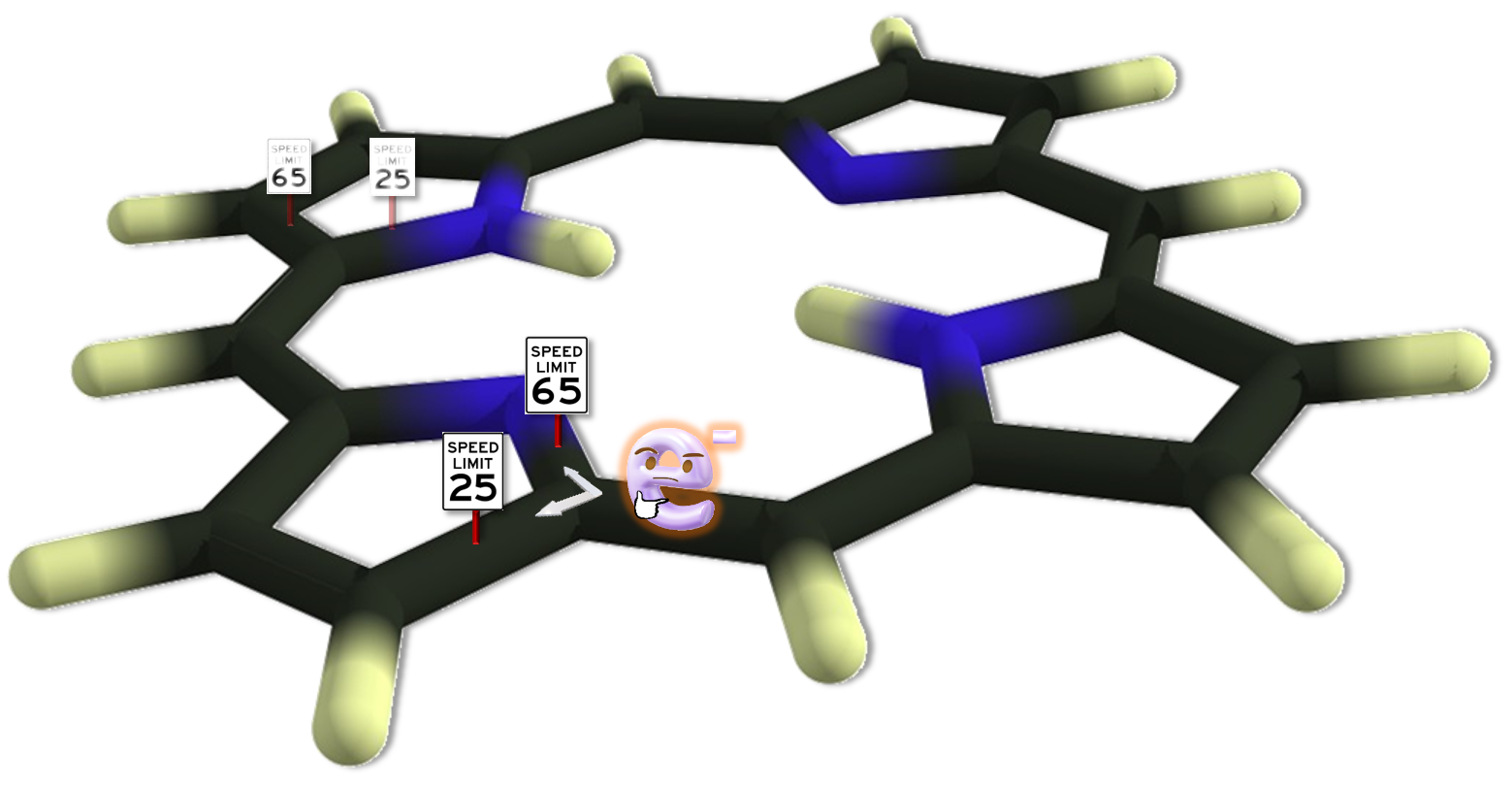
Aromaticity in Porphyrinoids
| Posted on March 12, 2018 at 8:45 AM |
The role of aromaticity in porphyrinoids is a current subject of debate due to the intricate structure of these macrocycles, which can adopt Hückel, Möbius and even figure-eight conformers. One of the main challenges in these large π-conjugated structures is identifying the most conjugated pathway because, among aromaticity descriptors, there are very few that can be applied coherently to this variety of conformers.
As a result of a joint colaboration with Miquel Torrent-Sucarrat, Mercedes Alonso and Julia Contreras-Garcia, we have recently published a paper on Phys. Chem. Chem. Phys. studying the most aromatic circuits in porphyrinoids. The main authors of the paper are Irene Casademont (PhD student at the UPV/EHU and the DIPC) and Tatiana Woller (PhD student at the Vrije Universiteit Brussel). In this work, we have found that two new electronic aromaticity indices AV1245 and AVmin developed in our group, provide a reliable description of the aromatic pathways in a series of nine porphyrinoids. Not many indices can be used to identify the most aromatic pathway in a macrocyclic (for instance, NICS which is probably the most popular index cannot be calculated for particular circuits) and in our study we have also used BLA, BOA, FLU and HOMA. All these indices agree on the general features of these compounds, such as the fulfillment of Hückel's rule or which compounds should be more or less aromatic from the series. However, only AVmin can identify the most aromatic circuit in all the molecules. Our results evince the difficulty of finding the most aromatic pathway in the macrocycle for large porphyrinoids.
We study the effect of the exchange in DFT functionals on the description of the aromaticity of the porphyrinoids. The amount of exact exchange quantitatively changes the picture for most aromaticity descriptors, AVmin being the only exception that shows the same qualitative results in all cases.
The paper has been published in Phys. Chem. Chem. Phys.
Tetrahalodiboranes
| Posted on February 20, 2018 at 7:10 AM |
Tetrahalodiboranes are enigmatic compounds, which are used for the doping of silicon with B+ ions for semiconductor device fabrication. Despite their lability and immense reactivity potential, the relative absence of tetrahalodiboranes is due to their difficult preparation, involving gas-phase synthetic steps. Recently, the first transition-metal complex of a diboranyl dianion has been prepared and characterized by the group of Prof. Braunschweig.
Our joint computational analysis with Prof. Óscar Jimenez-Halla suggests the presence of two multicenter PtB2 3c-2e bonds, similar to the two B3 3c- 2e bonds seen in Himmel’s rhomboidal B4 compound and the B2I4 unit can be seen as an olefin analogue. The compound represents the first example of intact coordination of B2X4 (X = halide) unit of any type to a metal center. These results provide a glimpse of the potentially exciting coordination chemistry of tetrahalodiboranes, about which very little is currently known.
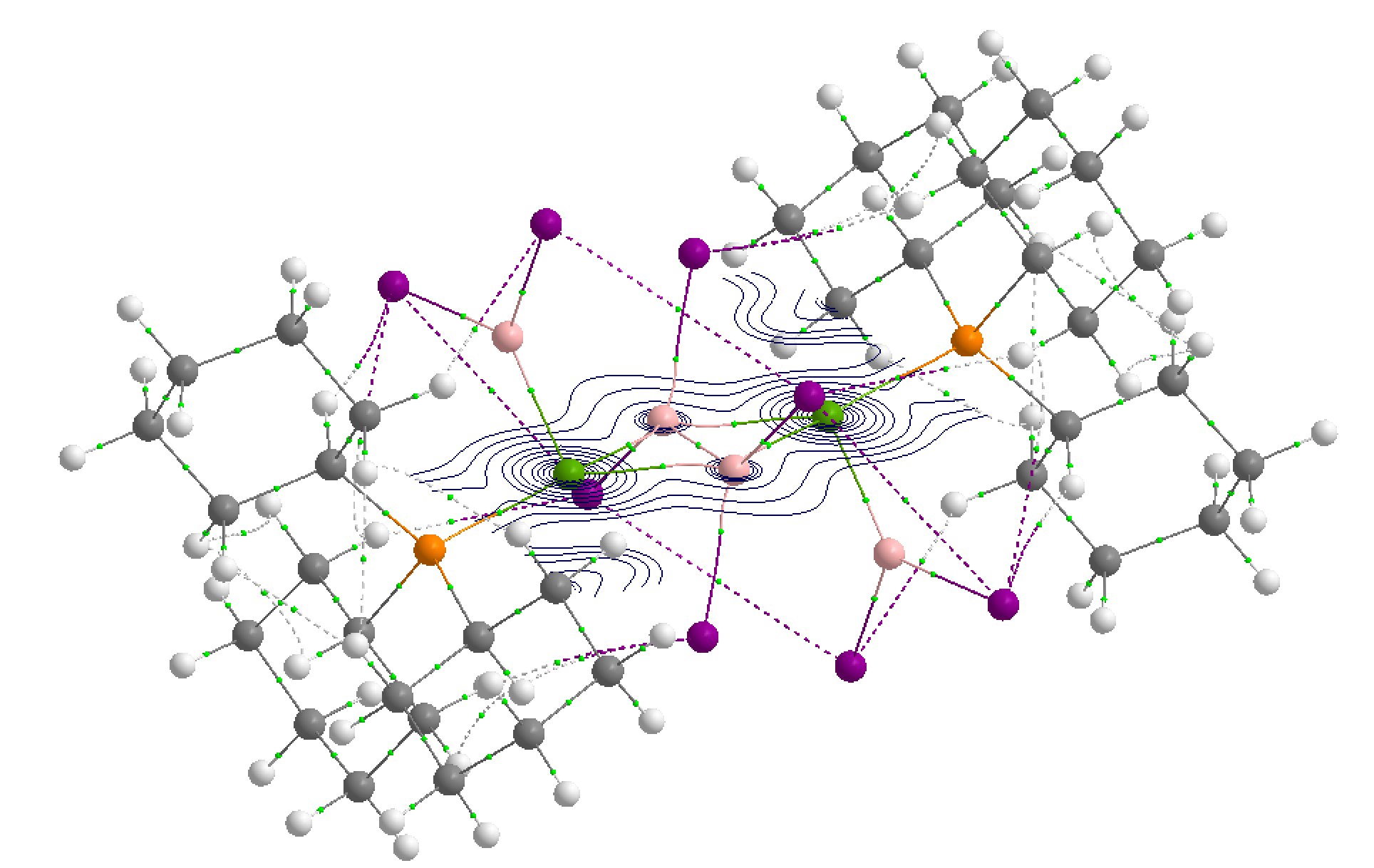
The full work has been published in Angew. Chemie Int. Ed.
A new fingerprint of weak molecular interactions
| Posted on November 11, 2017 at 10:05 AM |
The so-called van der Waals (vdW) interactions are one of the weakest forces in nature and yet they govern the stability of molecules and materials, having an essential role in molecular recognition, the stability of the double-helical structure of DNA and molecular adsorption processes on surfaces, amongst others. So far, only a universal relationship between the van der Waals energy and the distance between two atoms or molecules was known, being widely used to model these interactions in physics, chemistry, and biology.
In a recent work published in Phys. Rev. A., another universal signature of van der Waals interactions has been unveiled. This new fingerprint of weak molecular interactions is numerically more robust than the earlier condition on the energy and could thus provide a handy tool for the development of new methods to analyze the electronic structure of molecular systems. By means of perturbation theory, we have shown that the interelectronic part of the pair density, which is the workhorse of electronic structure methods and provides a distribution of the electron pairs in the space, decays as 1/R^3 for two molecular fragments separated by a distance R. The main author of this work is Mireia Via-Nadal (PhD student at the UPV/EHU and the DIPC), to which Mauricio Rodriguez-Mayorga has also contributed.
This result opens the door to produce density-dependent non-covalent interaction corrections in density and density matrix functional theories (DFT and DMFT). This possibility will be explored in our laboratory.
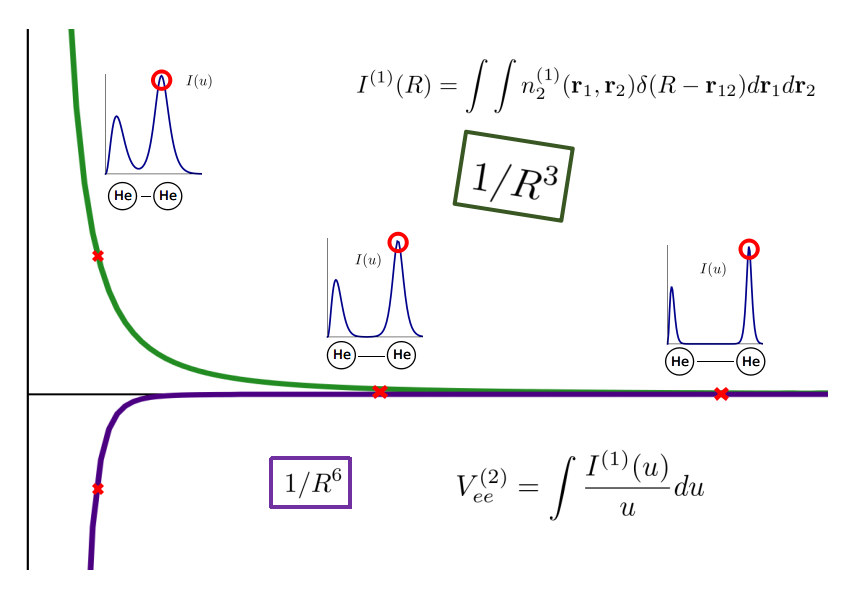
This work has been published in Phys. Rev. A.
Comprehensive benchmarking of DMFT approximations
| Posted on October 3, 2017 at 12:30 PM |
The energy usually serves as a yardstick in assessing the performance of approximate methods in computational chemistry. After all, these methods are mostly used for the calculation of the electronic energy of chemical systems. However, computational methods should be also aimed at reproducing other properties, such strategy leading to more robust approximations with a wider range of applicability.
In this work, we have suggested a battery of ten tests with the aim to analyze density matrix functional approximations (DMFAs), including several properties that the exact functional should satisfy. The tests are performed on a two-electron model system with varying electron correlation, carrying a very small computational effort. Our results not only put forward a complete and exhaustive benchmark test for DMFAs, currently lacking, but also reveal serious deficiencies of existing approximations that lead to important clues in the construction of more robust DMFAs. The main author of this work is Mauricio Rodríguez-Mayorga who received the help of several co-authors: Eloy Ramos-Cordoba, Mireia Via-Nadal and Mario Piris.
This work has been published in Phys. Chem. Chem. Phys.
A Real Space Account of Electron Correlation
| Posted on March 5, 2017 at 11:15 AM |
Last year we published the first of series a three papers dealing with the separation of dynamic (weak) and nondynamic (strong) correlation. In that work, we put forward a global correlation indicator based on natural orbital occupancies. Now, with Eloy Ramos-Cordoba we have extended this approach to account for real-space weak and strong correlation. By multiplying the orbital contributions to these correlation indicators by the corresponding natural orbitals, we produce a three-dimensional pictures of dynamic and nondynamic correlation.
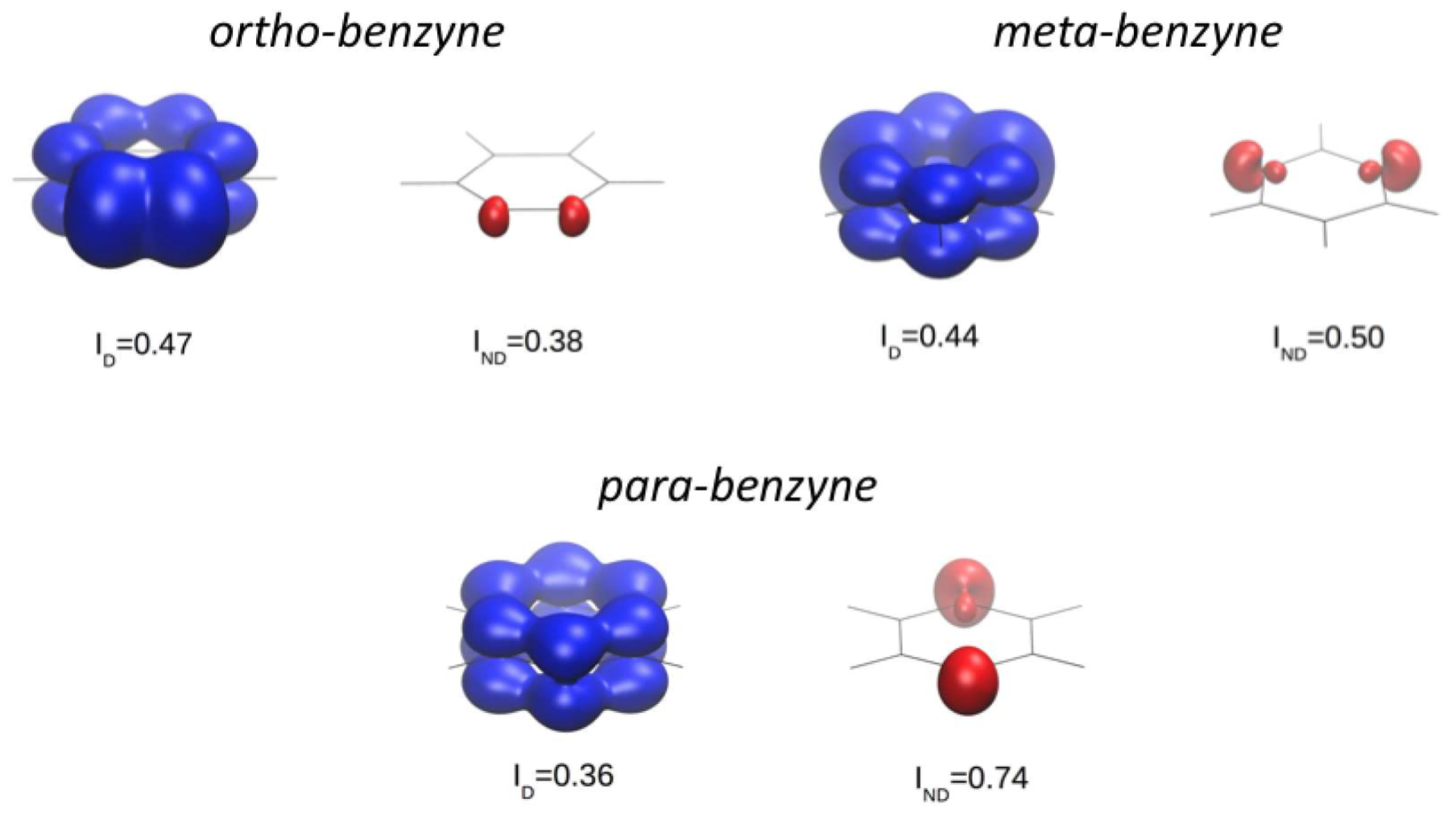
In the picture above we produce CASSCF pictures of dynamic and nondynamic correlation for the ortho-, meta-, and para-benzine singlet diradicals, which have two unpaired electrons. These unpaired electrons are easily located in the space using the real-space nondynamic correlation indicator suggested in this work.
This work has been published in J. Chem. Theory Comput.